VEXAS Syndrome: Every Ophthalmologist Must Know
In 2020, a landmark paper in the New England Journal of Medicine described 25 men with severe, treatment-resistant inflammation and a previously unrecognised genetic mutation — and changed how we think about adult-onset autoinflammatory disease forever (1). VEXAS syndrome is that disease. It sits at the crossroads of haematology and rheumatology, but the ophthalmologist is often the first specialist to encounter it. Knowing what to look for can save your patient years of diagnostic wandering.
What Is VEXAS, and Why Does It Happen?
VEXAS stands for Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic. It is caused by somatic mutations in the UBA1 gene located on the X chromosome (1). UBA1 encodes the E1 ubiquitin-activating enzyme, which is essential for the ubiquitin-proteasome pathway, the cell's main system for tagging defective proteins for degradation. When the mutation occurs in haematopoietic stem cells, it produces a catalytically deficient isoform called UBA1c, which accumulates in myeloid cells (neutrophils, monocytes) and drives a hyperinflammatory state (2). Critically, these mutations are acquired in adult life, they are not inherited. This is what makes VEXAS the first example of a monogenic autoinflammatory disease arising purely from somatic mosaicism.
Because UBA1 is X-linked, the condition predominantly affects men. The prevalence is approximately 1 in 4,269 males over the age of 50 (3). Women can be affected through X-chromosome monosomy (Turner syndrome) or skewed X-inactivation, though this is uncommon. The most frequently identified mutations affect the methionine at amino acid position 41 of UBA1 — specifically p.Met41Thr, p.Met41Val, and p.Met41Leu (2). The p.Met41Val variant carries the worst prognosis and the p.Met41Thr is the most commonly seen in patients with ocular involvement (4,5).
The Systemic Picture: A Master Mimic
The hallmark of VEXAS is recurrent, treatment-refractory inflammation affecting multiple organ systems. Fever occurs in about 65% of patients and weight loss in around 55% (2). Skin involvement is the most frequent finding, seen in up to 84% of cases — ranging from neutrophilic dermatosis and vasculitis to erythema nodosum and urticarial plaques (2). Relapsing polychondritis occurs in up to half of patients, presenting as auricular chondritis or nasal chondritis; a significant proportion of patients previously labelled as idiopathic relapsing polychondritis carry UBA1 mutations (6). Lung infiltrates, pleuritis, and venous thromboembolism are common. The haematological picture is equally striking: macrocytic anaemia (with normal B12 and folate), thrombocytopenia, lymphopenia, and myelodysplastic syndrome (MDS) in more than half of cases (2,7). The five-year mortality is 18–40% (8).
Because VEXAS mimics relapsing polychondritis, Sweet syndrome, polyarteritis nodosa, Behcet's disease, and seronegative spondylarthritis, patients are frequently managed under these diagnoses for months to years before the true cause is identified (2,9). The clue is refractoriness — a patient whose polychondritis or episcleritis never quite settles despite appropriate treatment, combined with unexplained macrocytosis or cytopenia, should raise immediate suspicion.
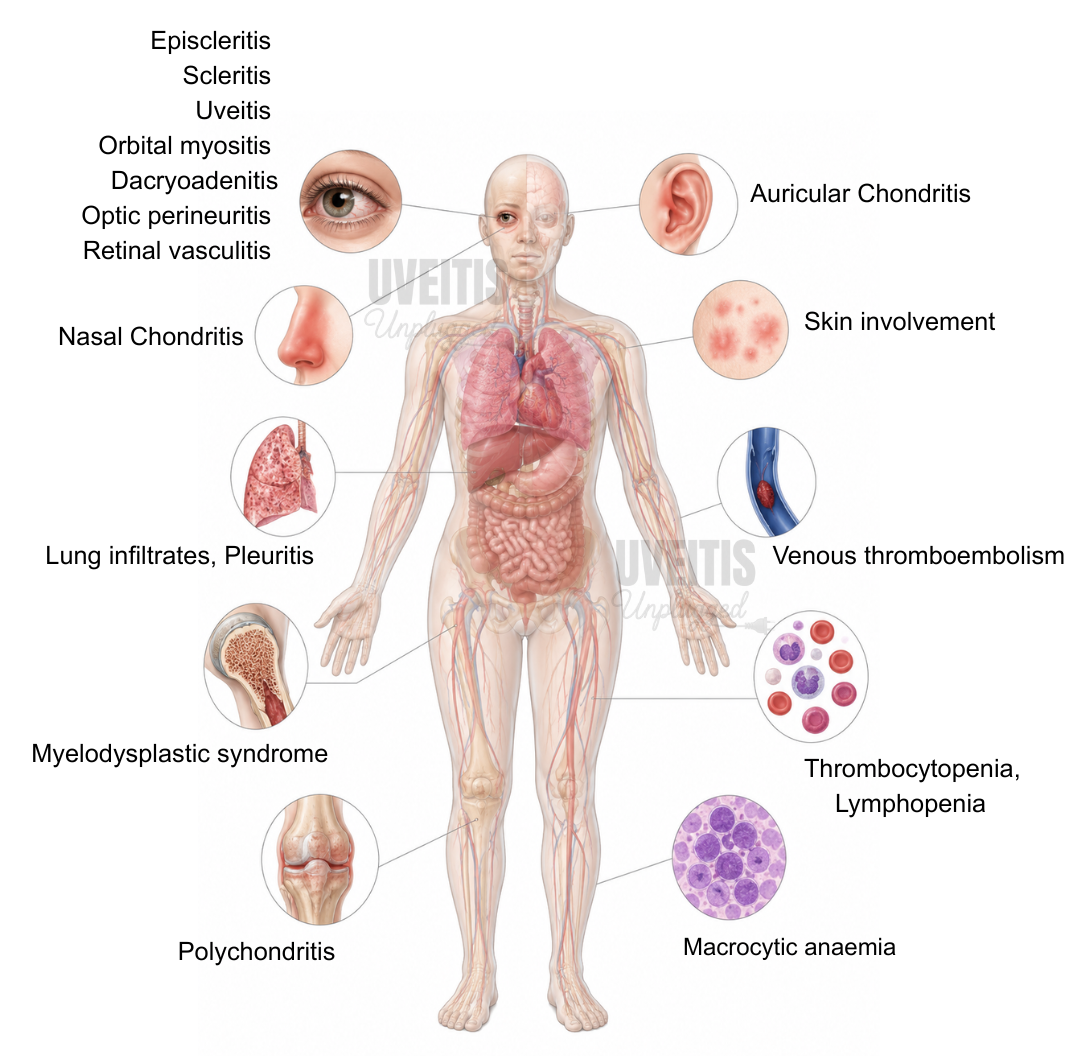
Ocular and Orbital Involvement
Ocular involvement is seen in up to 40% of VEXAS patients and may be the first presenting symptom of the syndrome (4,10). A recent systematic review and meta-analysis of 204 individuals with VEXAS found periorbital oedema to be the most common ocular manifestation (40.7%), followed by episcleritis (13.7%), scleritis (13.7%), and uveitis (12.3%) (10). Orbital myositis, dacryoadenitis, optic perineuritis, and retinal vasculitis have also been reported, though less frequently (1,4,10).
Orbital inflammation in VEXAS is characteristically recurrent and episodic, often self-limiting but prone to relapse. Cases of superior orbital fissure syndrome, dacryoadenitis, and proptosis secondary to idiopathic orbital inflammation have been described (11). Orbital myositis most frequently involves the medial and superior rectus muscles (12,13). Importantly, ocular symptoms may persist even when systemic inflammation appears to be under control — regular ophthalmological follow-up is therefore essential throughout the disease course (12,13).
Uveitis in VEXAS is typically anterior, although posterior manifestations including choroidal effusion, serous retinal detachment, and retinal vasculitis have been documented in severe cases (1,14). One notable case demonstrated VEXAS overlapping with Behcet's disease presenting with retinal uveitis (9). The coincidence of episcleritis with relapsing polychondritis should be a particular trigger to investigate for VEXAS (4,5).
Pearls to Decode VEXUS
- Think VEXAS in the steroid-refractory older male. If your patient has recurrent episcleritis, scleritis, or orbital inflammation that keeps coming back despite treatment, and is male and over 50, order a full blood count before anything else.
- Unexplained macrocytosis is a red flag. VEXAS frequently presents with macrocytic anaemia with normal B12, folate, and copper. An MCV > 100 fL in a man with ocular inflammation should prompt haematology referral.
- Episcleritis + relapsing polychondritis = test for UBA1. This combination in a middle-aged or elderly man has a high positive predictive value for VEXAS (4,5).
- Order UBA1 sequencing from peripheral blood. You do not need bone marrow to confirm the diagnosis — UBA1 genetic testing from peripheral blood is usually sufficient and is the gold standard (1,3).
- Orbital inflammation may recur after systemic control. Always arrange long-term ophthalmological follow-up even when rheumatology or haematology declares the disease well-controlled (12,13).
- Look at the bone marrow aspirate smear. If a bone marrow biopsy is done, look for cytoplasmic vacuolisation in myeloid and erythroid precursors — a threshold of ≥10% precursors with vacuoles has 100% sensitivity and specificity for the UBA1 mutation (2).
Treatment: Difficult, Evolving, and Hopeful
Systemic corticosteroids are the first-line treatment but flares are frequent when doses are tapered. Steroid-sparing agents including methotrexate, mycophenolate, and azathioprine have shown modest benefit (2,15). Among targeted agents, tocilizumab (anti-IL-6) and ruxolitinib (JAK1/2 inhibitor) have demonstrated the most consistent results. Ruxolitinib has been shown to be more effective than other JAK inhibitors, achieving clinical and laboratory response in over 80% of patients at 3 months in retrospective analyses (15). Azacitidine, a hypomethylating agent, is useful particularly when MDS co-exists (2). For refractory cases, allogeneic haematopoietic stem cell transplantation remains the only potentially curative option (2,8,15).
When using tocilizumab in patients with gastrointestinal involvement, caution is warranted — bowel perforation has been reported in a small number of VEXAS patients receiving this agent (2). Ophthalmically, orbital involvement may be episodic and self-limiting, but both systemic immunosuppression and targeted therapy are required when disease is persistent or vision-threatening.
Clinical Pearls for Busy Clinicians
- VEXAS is not rare — the prevalence among men over 50 is about 1 in 4,000, which means you will see it in a busy uveitis or inflammatory eye clinic (3).
- The p.Met41Thr mutation is the most common. It is also the most frequently associated with ocular involvement, particularly episcleritis and periorbital oedema (4,5).
- Diagnosis requires genetic testing. There are no specific autoantibodies for VEXAS — ANA, ANCA, RF, and ACE are all typically negative. The diagnosis cannot be made on clinical or radiological grounds alone.
- 5-year mortality is significant at 18–40%. These patients need multidisciplinary care with haematology, rheumatology, and ophthalmology working together (8).
- PET/CT can show diffuse bone marrow uptake. This is often one of the most consistent imaging findings in VEXAS and can alert you to the diagnosis when performed for fever of unknown origin (7).
REFERENCES:
- Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383(27):2628-2638. PMID: 33108101.
- Vitale A, Caggiano V, Bimonte A, et al. VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases. Intern Emerg Med. 2023;18(3):711-722. PMID: 36662353.
- Beck DB, Bodian DL, Shah V, et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA. 2023;329(4):318-324. PMID: 36633837.
- Fanlo P, Lopez de San Roman M, Fonollosa A, et al. Episcleritis and periorbital edema secondary to VEXAS syndrome. Arch Soc Esp Oftalmol. 2023;98(10):607-610. PMID: 37567469.
- Myint K, Patrao N, Vonica O, Vahdani K. Recurrent superior orbital fissure syndrome associated with VEXAS syndrome. J Ophthalmic Inflamm Infect. 2023;13(1):39. PMID: 37695478.
- Khitri MY, Guedon AF, Georgin-Lavialle S, et al. Comparison between idiopathic and VEXAS-relapsing polychondritis: analysis of a French case series of 95 patients. RMD Open. 2022;8(2):e002255. PMID: 35853682.
- Lohaus N, Schaab J, Schaer D, Balabanov S, Huellner MW. VEXAS syndrome with tracheal involvement but absence of vasculitis in FDG PET/CT. Clin Nucl Med. 2023;48(9):e444-e445. PMID: 37494583.
- Kotter I, Krusche M. VEXAS syndrome: an adult-onset autoinflammatory disorder with underlying somatic mutation. Curr Opin Rheumatol. 2025;37(1):21-31. PMID: 39470174.
- Matsumoto H, Asano T, Tsuchida N, et al. Behcet's disease with a somatic UBA1 variant: expanding spectrum of autoinflammatory phenotypes of VEXAS syndrome. Clin Immunol. 2022;238:108996. PMID: 35395395.
- Quigley C, Pietris J, Ang T, Zgaga L, Selva D. Ocular features of VEXAS syndrome: a systematic review and meta-analysis. Am J Ophthalmol. 2025;276:50-63. PMID: 40157447.
- Lokhande A, Jarmale S, Vaishnav YJ, Schaefer J. An orbital manifestation of VEXAS syndrome. Ophthalmic Plast Reconstr Surg. 2023. PMID: 37256692.
- Beecher M, Tong JY, Halliday LA, Hissaria P, Selva D. Recurrent orbital inflammation associated with VEXAS syndrome. Orbit. 2022;1-4. PMID: 35730610.
- Topilow JS, Ospina Cardona D, Beck DB, et al. Novel genetic mutation in myositis-variant of VEXAS syndrome. Rheumatology (Oxford). 2022;61(12):e371-e373. PMID: 35560193.
- Tozaki N, Tawada C, Niwa H, et al. A case of VEXAS syndrome with decreased oxidative stress levels after oral prednisone and tocilizumab treatment. Front Med. 2022;9:1046820. PMID: 36530945.
- Sujobert P, Heiblig M, Jamilloux Y. VEXAS: where do we stand 2 years later? Curr Opin Hematol. 2023;30(2):64-69. PMID: 36728604.
- Abumanhal M, Leibovitch I, Zisapel M, et al. Ocular and orbital manifestations in VEXAS syndrome. Eye (Lond). 2024;38(9):1748-1754. PMID: 38548942.
